211service.com
Primera simulación de nivel atómico de una batería completa
Cuando se trata de desarrollar la próxima generación de tecnología, el mayor cuello de botella es posiblemente la batería. Los ingenieros necesitan mejores baterías para los vehículos eléctricos, para el almacenamiento de energía en las redes eléctricas y, por supuesto, para los dispositivos electrónicos de consumo.
Estas baterías deben entregar una corriente más alta durante más ciclos de descarga con una mayor densidad de energía, por nombrar solo algunos de los desafíos.
Construir y probar nuevos diseños de baterías requiere mucho tiempo, es difícil y costoso. Por lo tanto, es útil para los electroquímicos simular el rendimiento de una batería antes de ensuciarse las manos.
Eso es complicado. Nadie ha podido simular una batería completa a nivel atómico debido a la complejidad de los procesos que se llevan a cabo y las limitaciones de las técnicas de modelado actuales.
Hoy eso cambia gracias al trabajo de Wolf Dapp en el Instituto de Simulación Avanzada y Martin Muser en la Universidad de Saarlandes, ambos en Alemania. Estos chicos han simulado el comportamiento de una batería completa a escala atómica. Y su simulación reproduce muchas de las características reales de una batería desde los primeros principios por primera vez.
En los últimos años, los científicos informáticos han logrado un progreso significativo en la simulación de varios aspectos del comportamiento de la batería. Estos modelos funcionan en la mesoescala, más pequeños que los electrodos pero más grandes que las moléculas. Las simulaciones se basan en datos experimentales para modelar cosas como conductividad iónica y electrónica, coeficientes de difusión, densidades de corriente, potenciales electroquímicos, etc.
Estos modelos tienen un serio inconveniente: tienen poco poder de predicción cuando se trata de nuevos materiales o combinaciones de materiales para los que no se dispone de datos experimentales. Para predecir el comportamiento de nuevos materiales, los electroquímicos necesitan modelar baterías en la escala de átomos y moléculas.
Eso es difícil porque las técnicas que utilizan los científicos informáticos para simular el comportamiento de los átomos y las moléculas no son adecuadas para las baterías. Estas simulaciones están diseñadas para sistemas que están en equilibrio o cerca de él. Funcionan igualando el potencial químico o minimizando la energía del sistema. Sin embargo, la diferencia en el potencial químico entre dos electrodos es precisamente lo que impulsa el transporte de carga en una batería, dicen Dapp y Muser.
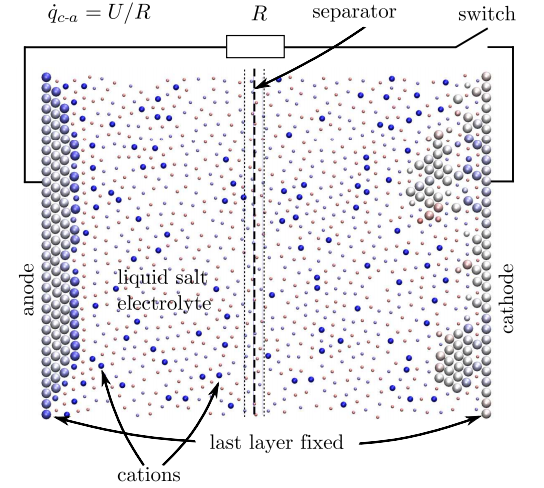
Entonces, para modelar una batería en su conjunto, el modelo de computadora debe tener en cuenta cualquier cambio en el potencial energético o químico en cada paso del cálculo. Esto es exactamente lo que han hecho Dapp y Muser. En su modelo, la carga es una variable que se puede intercambiar entre átomos y enlaces en cada etapa del cálculo.
Las simulaciones resultantes son pequeñas pero impresionantes. Su nanobatería consta de 358 átomos, de los cuales 118 forman los electrodos. El cátodo se cubre inicialmente con una capa de 20 átomos con 39 átomos ionizados positivamente disueltos en el electrolito.
Luego, el cálculo procede en pasos en los que los átomos pueden moverse e intercambiar carga a medida que el sistema evoluciona. La simulación completa consta de unos 10 millones de estos pasos.
Los resultados son notables porque en realidad reproducen las curvas de descarga genéricas de baterías macroscópicas reales. Por ejemplo, una temperatura de funcionamiento más baja reduce la capacidad efectiva de la batería simulada. Y lo más importante de todo, la simulación reproduce la forma en que se gastan las baterías normales. Tras la recarga, el rendimiento de la batería se degrada ligeramente y la morfología de la superficie del electrodo cambia durante el funcionamiento de la batería, dicen Dapp y Muser.
Estos muchachos dicen que el trabajo en esta etapa es solo un modelo de prueba de principio y que hay varias formas en las que se puede mejorar en el futuro. Por ejemplo, modelan el electrolito utilizando partículas que tienen una carga fija y, por lo tanto, no pueden intercambiarla.
No es así como funcionan los electrolitos en baterías reales, por lo que este es un defecto importante del nuevo método. Pero Dapp y Muser pretenden corregir esto. Esta idealización será abandonada en trabajos futuros, dicen.
En general, este parece ser un trabajo importante. Este tipo de modelo podría mejorar drásticamente el poder predictivo de las simulaciones de baterías y, por lo tanto, ayudaría a ahorrar a los electroquímicos una cantidad significativa de tiempo, energía y dinero antes de comenzar experimentos detallados.
El resultado final debería ser mejores baterías, pero queda un largo camino por recorrer antes de eso.
Ref: arxiv.org/abs/1308.3424 : Reacciones redox con potenciales empíricos: Simulaciones de descarga de batería atomística