211service.com
Medir la forma de las proteínas ahora es más fácil gracias a las matemáticas
Estructura de la proteína Los análisis de los núcleos de proteínas revelan diferencias fundamentales entre la solución y las estructuras cristalinas
Las proteínas son los componentes básicos de la vida. Son largas cadenas de aminoácidos que se autoensamblan en máquinas moleculares de extraordinaria complejidad. Estas máquinas incluyen dispositivos informáticos llamados ribosomas, esqueletos de andamios llamados microtúbulos y máquinas para caminar con forma de piernas llamadas kinesina, entre muchas otras.
Este proceso de autoensamblaje es una de las grandes maravillas de la ciencia moderna, casi como si una cadena de piezas de Lego se ensamblara repentinamente en un robot. Nadie está seguro de cómo sucede esto, pero los científicos saben que la forma de la estructura resultante determina la función de la proteína y cómo interactúa con otras proteínas.
Por lo tanto, medir la forma de las proteínas es una tarea crucial. El método más común es formar cristales a partir de proteínas y luego usar cristalografía de rayos X para determinar la estructura de la proteína.
Eso es un problema porque la mayoría de las proteínas no forman cristales. E incluso cuando lo hacen, es posible que no todas las moléculas de proteína tomen la misma forma a medida que se empaquetan, lo que genera imprecisiones.
Otra técnica, llamada resonancia magnética nuclear, crea imágenes de proteínas en solución, pero requiere que estén muy juntas en paquetes. Nuevamente, solo unas pocas proteínas pueden hacer esto.
Sin embargo, una pequeña fracción de proteínas se puede visualizar con ambas técnicas. Eso es útil porque permite a los biólogos moleculares comparar las estructuras que produce cada técnica.
Y resulta que las estructuras encontradas por cada técnica difieren de manera significativa. Pero no está claro exactamente qué causa las diferencias y cómo interpretarlas.
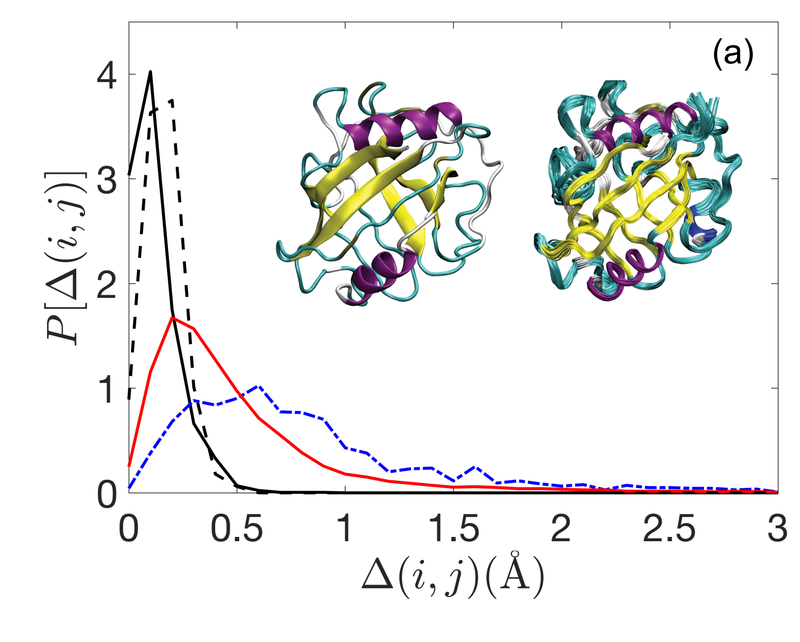
Hoy eso cambia, al menos en parte, gracias al trabajo de Zhe Mei y sus colegas de la Universidad de Yale en New Haven. Este equipo ha medido la diferencia en la estructura de la proteína determinada por cristalografía de rayos X y RMN. Y han descubierto por qué surge la diferencia y cómo corregirla.
El equipo comenzó compilando una base de datos de proteínas con estructuras determinadas por ambas técnicas en alta resolución. Esa resulta ser una lista relativamente pequeña: solo 16 proteínas cumplen los requisitos.
Los investigadores también crearon una base de datos de estructuras de proteínas de cristalografía de rayos X determinadas por varios grupos diferentes a diferentes temperaturas. Esto permitió al equipo estudiar cómo la temperatura influye en la estructura.
Luego crearon un modelo matemático de la forma en que las proteínas se empaquetan para formar cristales sólidos o paquetes en solución para RMN.
Resulta que la densidad de empaquetamiento puede explicar exactamente la diferencia en la estructura de ambas técnicas, los paquetes en solución tienen una densidad más alta que los cristales. Identificamos la base física de estas diferencias mediante el modelado de núcleos de proteínas como paquetes atascados de partículas con forma de aminoácido, dicen los investigadores.
También pueden afinar su modelo matemático variando la energía térmica utilizada para generar los empaques. De hecho, el empaquetamiento de proteínas que no está influenciado por la temperatura tiene aproximadamente la misma densidad que las estructuras determinadas por cristalografía de rayos X.
Esto sugiere que la temperatura juega un papel importante en la estructura de empaquetamiento de proteínas, ya que las estructuras determinadas por RMN son más densas. Estos resultados indican que los sistemas termalizados pueden empacar más densamente que los sistemas atérmicos, lo que sugiere una base física para las diferencias estructurales entre las estructuras de proteínas resueltas por RMN y cristalografía de rayos X, dicen Mei y compañía.
Sin embargo, la temperatura no es toda la historia. Las proteínas en las estructuras cristalinas se ven obligadas a adoptar una determinada forma, y esto reduce la cantidad de distorsión térmica que puede sufrir la molécula.
Entonces, el resultado de Mei y compañía plantea una pregunta interesante: ¿En qué medida la estructura de la proteína es el resultado de la temperatura o el resultado del empaquetamiento de cristales?
Eso tendrá que esperar para más trabajo.
Ref: arxiv.org/abs/1907.08233 : Los análisis de los núcleos de proteínas revelan diferencias fundamentales entre la solución y las estructuras cristalinas