211service.com
Las células inmunes rebeldes que destruyen el cerebro
En los primeros años de su carrera en la investigación del cerebro, Beth Stevens pensaba en la microglía con molestia, si es que pensaba en ella. Cuando miró a través de un microscopio y vio estas células ubicuas con sus tentáculos de araña, hizo lo que la mayoría de los neurocientíficos habían estado haciendo durante generaciones: miró más allá de ellos y se concentró en el resto del tejido cerebral, tal como se puede mirar a través de motas de suciedad en un parabrisas.
¿Que están haciendo alli? pensó. Están en el camino.

Esta historia fue parte de nuestra edición de mayo de 2016
- Ver el resto del número
- Suscribir
Stevens nunca hubiera imaginado que solo unos años más tarde, estaría dirigiendo una laboratorio en Harvard y Boston's Children's Hospital dedicado al estudio de estos pequeños grupos oscuros. O que estaría argumentando en las principales revistas científicas del mundo que la microglía podría ser la clave para comprender no solo el desarrollo normal del cerebro, sino también las causas del Alzheimer, la enfermedad de Huntington, el autismo, la esquizofrenia y otros trastornos cerebrales intratables.
La microglía es parte de una clase más grande de células, conocidas colectivamente como glía, que realizan una serie de funciones en el cerebro, guían su desarrollo y sirven como su sistema inmunológico al engullir células enfermas o dañadas y transportar desechos. Junto con su frecuente colaborador y mentor, el biólogo de Stanford Ben Barres, y un grupo cada vez mayor de otros científicos, Stevens, de 45 años, está demostrando que estas células ignoradas durante mucho tiempo son más que meros trabajadores de apoyo para las neuronas que rodean. Su trabajo ha planteado una sugerencia provocativa: que los trastornos cerebrales de alguna manera podrían ser desencadenados por nuestras propias defensas corporales que se estropean.
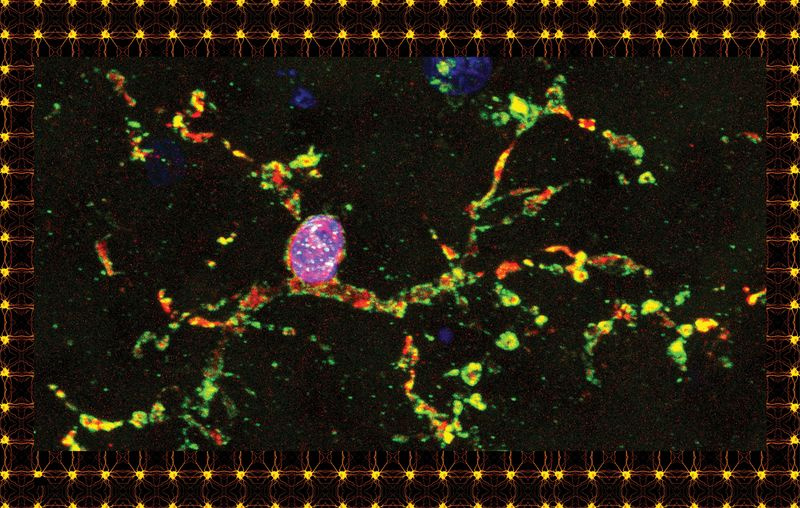
Inicio de página: Una célula microglial de un cerebro humano, teñida con fines de investigación. Arriba: un tipo de célula glial conocida como oligodendrocitos.
En un innovador papel En enero, Stevens e investigadores del Instituto Broad del MIT y Harvard demostraron que la microglía aberrante podría desempeñar un papel en la esquizofrenia, causando o al menos contribuyendo a la pérdida masiva de células que puede dejar a las personas con devastadores defectos cognitivos. Crucialmente, los investigadores señalaron una vía química que podría ser el objetivo de retrasar o detener la enfermedad. La semana pasada, Stevens y otros investigadores publicado un hallazgo similar para el Alzheimer.
Esto podría ser solo el comienzo. Stevens también está explorando la conexión entre estas diminutas estructuras y otras enfermedades neurológicas, trabajo que le valió $625,000 Beca de genio de la Fundación MacArthur septiembre pasado.
Todo esto plantea preguntas intrigantes. ¿Es posible que muchos trastornos cerebrales comunes, a pesar de la amplia variedad de síntomas, sean causados o al menos empeorados por el mismo culpable, un componente del sistema inmunitario? Si es así, ¿podrían tratarse muchos de estos trastornos de manera similar, deteniendo estas células rebeldes?
Maquinaria compleja
No sorprende que los científicos hayan ignorado durante años la microglía y otras células gliales en favor de las neuronas. Las neuronas que se disparan juntas nos permiten pensar, respirar y movernos. Vemos, oímos y sentimos usando neuronas, y formamos recuerdos y asociaciones cuando las conexiones entre diferentes neuronas se fortalecen en las uniones entre ellas, conocidas como sinapsis. Muchos neurocientíficos argumentan que las neuronas crean nuestra propia conciencia.
Glia, por otro lado, siempre se ha considerado menos importante e interesante. Tienen deberes de peatones, como suministrar nutrientes y oxígeno a las neuronas, así como limpiar los productos químicos perdidos y acarrear la basura.
Los científicos saben acerca de la glía desde hace algún tiempo. En la década de 1800, el patólogo Rudolf Virchow notó la presencia de pequeñas células redondas que llenaban los espacios entre las neuronas y las denominó neuralnkitt o neuroglia, que puede traducirse como masilla o pegamento para nervios. Una variedad de estas células, conocida como astrocitos, se definió en 1893. Y luego, en la década de 1920, el científico español Pio del Río Hortega desarrolló formas novedosas de teñir células extraídas del cerebro. Esto lo llevó a identificar y nombrar dos tipos más de células gliales, incluida la microglía, que son mucho más pequeñas que las demás y se caracterizan por su forma de araña y múltiples ramificaciones. Sugirió que solo cuando el cerebro se daña en la edad adulta, la microglía cobra vida y se precipita hacia la lesión, donde se pensaba que ayudaba a limpiar el área al comer células dañadas y muertas. Los astrocitos también aparecían a menudo en escena; se pensaba que creaban tejido cicatricial.
Esta convergencia de emergencia de microglia y astrocitos se denominó gliosis, y cuando Ben Barres ingresó a la escuela de medicina a fines de la década de 1970, estaba bien establecida como un sello distintivo de enfermedades neurodegenerativas, infecciones y una amplia gama de otras afecciones médicas. Pero nadie parecía entender por qué ocurrió. Eso intrigó a Barres, entonces un neurólogo en formación, que lo vio cada vez que miraba bajo un microscopio el tejido neural en peligro. Fue realmente fascinante, dice. El gran misterio era: ¿cuál es el sentido de esta gliosis? ¿Esta bien? ¿Es mala? ¿Está impulsando el proceso de la enfermedad o está tratando de reparar el cerebro lesionado?
Barres comenzó a buscar la respuesta. Aprendió cómo hacer crecer células gliales en un plato y aplicarles una nueva técnica de registro. Podía medir sus cualidades eléctricas, que determinan la señalización bioquímica que utilizan todas las células cerebrales para comunicarse y coordinar la actividad.
Desde el momento en que comencé a registrar las células gliales, pensé: '¡Oh, Dios mío!', recuerda Barres. La actividad eléctrica era más dinámica y compleja de lo que nadie había pensado. Estas extrañas propiedades eléctricas solo podrían explicarse si las células gliales estuvieran sintonizadas con las condiciones que las rodean y con las señales emitidas por las neuronas cercanas. Las células gliales de Barres, en otras palabras, tenían toda la maquinaria necesaria para entablar un diálogo complejo con las neuronas y, presumiblemente, para responder a diferentes tipos de condiciones en el cerebro.
Sin embargo, ¿por qué necesitarían esta maquinaria si simplemente estuvieran involucrados en la limpieza de células muertas? ¿Qué podrían estar haciendo? Resulta que en ausencia de sustancias químicas liberadas por la glía, las neuronas cometieron la versión bioquímica del suicidio. Barres también mostró que los astrocitos parecían desempeñar un papel crucial en la formación de sinapsis, las conexiones microscópicas entre las neuronas que codifican la memoria. Aisladas, las neuronas eran capaces de formar los apéndices espinosos necesarios para alcanzar las sinapsis. Pero sin astrocitos, eran incapaces de conectarse entre sí.
Casi nadie le creyó. Cuando era un joven miembro de la facultad de Stanford en la década de 1990, una de sus solicitudes de subvención a los Institutos Nacionales de Salud fue rechazada siete veces. Los revisores seguían diciendo: 'No, no hay forma de que la glía pueda estar haciendo esto', recuerda Barres. E incluso después de que publicamos dos artículos en Ciencias mostrando que [los astrocitos] tenían efectos profundos, casi de todo o nada en el control de la formación de sinapsis o la actividad de las sinapsis, ¡todavía no pude obtener financiación! Creo que todavía es difícil hacer que la gente piense en la glía como algo activo en el sistema nervioso.
Marcado para la eliminación
Beth Stevens llegó a estudiar la glía por accidente. Después de graduarse de la Universidad Northeastern en 1993, siguió a su futuro esposo a Washington, D.C., donde él consiguió trabajo en el Senado de los Estados Unidos. Stevens había estudiado medicina en la universidad y esperaba trabajar en un laboratorio de los Institutos Nacionales de Salud. Pero sin experiencia previa en investigación, fue rechazada rotundamente. Así que tomó un trabajo de camarera en un restaurante Chili's en las cercanías de Rockville, Maryland, y se presentó en los NIH con su currículum todas las semanas.
Después de unos meses, Stevens recibió una llamada de un investigador llamado Doug Fields, que necesitaba ayuda en su laboratorio. Fields estaba estudiando las complejidades del proceso en el que las neuronas se aíslan en una capa llamada mielina. Ese aislamiento es fundamental para la transmisión de los impulsos eléctricos.
Mientras Stevens pasó los años siguientes buscando un doctorado en la Universidad de Maryland, estaba intrigada por el papel que desempeñaban las células gliales en el aislamiento de las neuronas. En el camino, se familiarizó con otras ideas sobre las células gliales que comenzaban a surgir, especialmente del laboratorio de Ben Barres. Por eso, poco después de completar su doctorado en 2003, Stevens se encontró con un posdoctorado en el laboratorio de Barres en Stanford, a punto de hacer un descubrimiento crucial.
El grupo de Barres había comenzado a identificar los compuestos específicos que secretaban los astrocitos y que parecían hacer que las neuronas desarrollaran sinapsis. Y finalmente, notaron que estos compuestos también estimulaban la producción de una proteína llamada C1q.
La sabiduría convencional sostenía que C1q se activaba solo en las células enfermas (la proteína las marcaba para que las células inmunitarias las consumieran) y solo fuera del cerebro. Pero Barres lo había encontrado en el cerebro. Y fue en neuronas sanas que posiblemente se encontraban en su etapa más robusta: en el desarrollo temprano. ¿Qué estaba haciendo allí la proteína C1q?
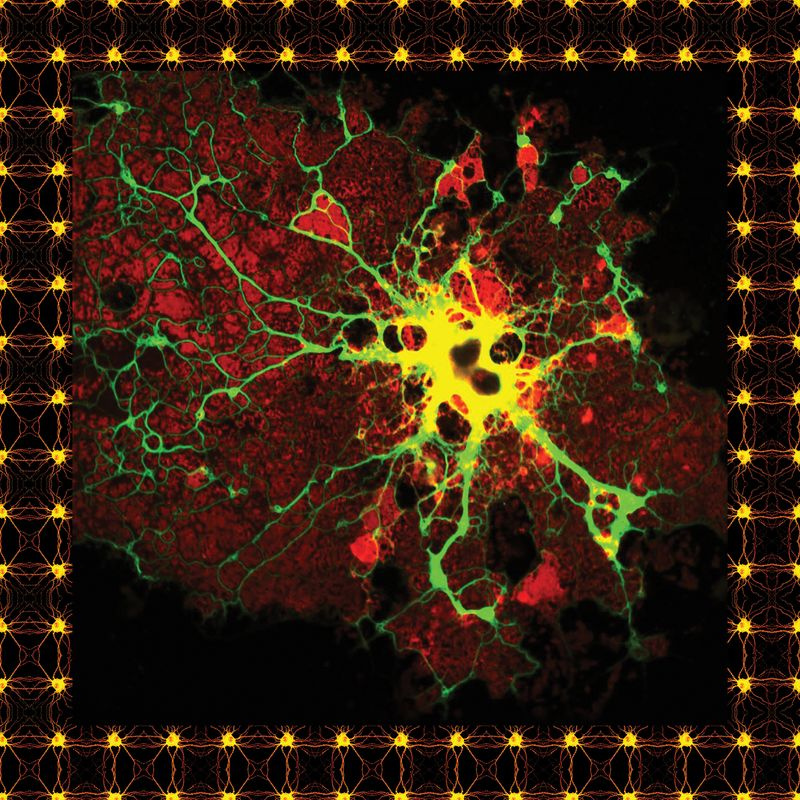
Un astrocito teñido.
La respuesta radica en el hecho de que marcar las células para su eliminación no es algo que suceda solo en cerebros enfermos; también es esencial para el desarrollo. A medida que se desarrolla el cerebro, sus neuronas forman muchas más conexiones sinápticas de las que eventualmente necesitarán. Solo los que se usan pueden permanecer. Esta poda permite el flujo más eficiente de transmisiones neuronales en el cerebro, eliminando el ruido que podría enturbiar la señal.
Pero se desconocía cómo funcionaba exactamente el proceso. ¿Era posible que C1q ayudara a indicar al cerebro que redujera las sinapsis no utilizadas? Stevens centró su investigación posdoctoral en averiguarlo. Podríamos haber estado completamente equivocados, recuerda. Pero fuimos a por ello.
Valió la pena. En un artículo de 2007, Barres y Stevens demostraron que C1q de hecho juega un papel en la eliminación de neuronas innecesarias en el cerebro en desarrollo. Y descubrieron que la proteína está prácticamente ausente en las neuronas adultas sanas.
Ahora los científicos se enfrentaban a un nuevo rompecabezas. ¿Aparece C1q en las enfermedades cerebrales porque el mismo mecanismo involucrado en la poda de un cerebro en desarrollo luego falla? De hecho, ya estaba aumentando la evidencia de que uno de los primeros eventos en enfermedades neurodegenerativas como el Alzheimer, el Parkinson y el Huntington fue la pérdida significativa de sinapsis.
Finalmente podríamos perseguir enfermedades que han estado sin control durante generaciones.
Cuando Stevens y Barres examinaron ratones criados para desarrollar glaucoma, una enfermedad neurodegenerativa que mata las neuronas en el sistema óptico, encontraron que C1q apareció mucho antes que cualquier otro signo detectable de que la enfermedad se estaba afianzando. Surgió incluso antes de que las células comenzaran a morir.
Esto sugirió que las células inmunitarias podrían, de hecho, causar la enfermedad, o al menos acelerarla. Y eso ofrecía una posibilidad intrigante: que se pudiera hacer algo para detener el proceso. Barres fundó una empresa, Annexon Biosciences, para desarrollar medicamentos que pudieran bloquear C1q. El artículo de la semana pasada publicado por Barres, Stevens y otros investigadores muestra que un compuesto que está siendo probado por Annexon parece ser capaz de prevenir la aparición de la enfermedad de Alzheimer en ratones criados para desarrollar la enfermedad. Ahora la compañía espera probarlo en humanos en los próximos dos años.
Caminos a los tratamientos
Para comprender mejor el proceso que C1q ayuda a desencadenar, Stevens y Barres querían descubrir qué es lo que realmente juega el papel de Pac-Man, devorando las sinapsis marcadas para la muerte. Era bien sabido que los glóbulos blancos conocidos como macrófagos engullían células enfermas e invasores extraños en el resto del cuerpo. Pero los macrófagos no suelen estar presentes en el cerebro. Para que su teoría funcionara, tenía que haber algún otro mecanismo. Y investigaciones posteriores han demostrado que las células que comen, incluso en cerebros sanos, son esos misteriosos grupos de material que Beth Stevens, durante años, había estado observando en el microscopio: la microglía que Río Hortega identificó hace casi 100 años.
Ahora, el laboratorio de Stevens en Harvard, que abrió en 2008, dedica la mitad de sus esfuerzos a descubrir qué están haciendo las microglías y qué las hace hacerlo. Resulta que estas células aparecen en el embrión de ratón en el día ocho, antes que cualquier otra célula cerebral, lo que sugiere que podrían ayudar a guiar el resto del desarrollo del cerebro y podrían contribuir a una serie de enfermedades del neurodesarrollo cuando salen mal.
Mientras tanto, también está ampliando su estudio sobre la forma en que diferentes sustancias determinan lo que sucede en el cerebro. C1q es en realidad solo la primera de una serie de proteínas que se acumulan en las sinapsis marcadas para su eliminación. Stevens ha comenzado a descubrir evidencia de que existe una amplia gama de moléculas protectoras de no me comas también. Es el equilibrio entre todas estas señales lo que regula si se convoca a la microglía para destruir las sinapsis. Los problemas en cualquiera podrían, posiblemente, estropear el sistema.
Cada vez hay más pruebas de que la microglía está involucrada en varios problemas psiquiátricos y del neurodesarrollo. El vínculo potencial con la esquizofrenia que se reveló en enero surgió después de que los investigadores del Instituto Broad, dirigidos por Steven McCarroll y un estudiante graduado llamado Aswin Sekar, siguieran un rastro de pistas genéticas que los condujo directamente al trabajo de Stevens. En 2009, tres consorcios de todo el mundo publicaron artículos que comparaban el ADN en personas con y sin esquizofrenia. Fue Sekar quien identificó un posible patrón: cuanto más un tipo específico de proteína estaba presente en las sinapsis, mayor era el riesgo de desarrollar la enfermedad. La proteína, C4, estaba estrechamente relacionada con C1q, la primera identificada en el cerebro por Stevens y Barres.
McCarroll sabía que la esquizofrenia ataca al final de la adolescencia y al principio de la edad adulta, una época en la que los circuitos cerebrales de la corteza prefrontal se someten a una poda extensa. Otros habían descubierto que las áreas de la corteza prefrontal se encuentran entre las más devastadas por la enfermedad, lo que conduce a una pérdida masiva de sinapsis. ¿Podría ser que la poda excesiva por parte de la microglía rebelde sea parte de lo que causa la esquizofrenia?
Para averiguarlo, Sekar y McCarroll se pusieron en contacto con Stevens y los dos laboratorios comenzaron a celebrar reuniones semanales conjuntas. Pronto demostraron que C4 también tenía un papel en la poda de sinapsis en el cerebro de ratones jóvenes, lo que sugiere que los niveles excesivos de la proteína podrían conducir a una poda excesiva y al adelgazamiento del tejido cerebral que parece ocurrir como síntomas como los episodios psicóticos empeoran.
Si el daño cerebral que se observa en el Parkinson y el Alzheimer proviene de una poda excesiva que podría comenzar temprano en la vida, ¿por qué los síntomas de esas enfermedades no aparecen hasta más tarde? Barres cree que lo sabe. Señala que el cerebro normalmente puede compensar una lesión reconectándose y generando nuevas sinapsis. También contiene mucha redundancia. Eso explicaría por qué los pacientes con enfermedad de Parkinson no muestran síntomas perceptibles hasta que han perdido el 90 por ciento de las neuronas que producen dopamina.
También podría significar que los síntomas sutiles podrían, de hecho, detectarse mucho antes. Barres apunta a un estudio de monjas publicado en 2000. Cuando los investigadores analizaron los ensayos que las monjas habían escrito al ingresar a sus conventos décadas antes, descubrieron que las mujeres que desarrollaron la enfermedad de Alzheimer habían mostrado menos densidad de ideas incluso en sus 20 años. Creo que la implicación de eso es que podrían ser enfermedades de por vida, dice Barres. El proceso de la enfermedad podría continuar durante décadas y el cerebro solo está compensando, reconectando, creando nuevas sinapsis. En algún momento, la microglía se activa para eliminar demasiadas células, argumenta Barres, y los síntomas de la enfermedad comienzan a manifestarse por completo.
Convertir esta idea en un tratamiento está lejos de ser sencillo, porque aún queda mucho por aclarar. Quizás una respuesta demasiado agresiva de la microglía esté determinada por alguna combinación de variantes genéticas que no todos comparten. Stevens también señala que enfermedades como la esquizofrenia no son causadas por una mutación; más bien, una amplia gama de mutaciones con pequeños efectos causan problemas cuando actúan en concierto. Los genes que controlan la producción de C4 y otras proteínas del sistema inmunitario pueden ser solo una parte de la historia. Eso puede explicar por qué no todas las personas que tienen una mutación C4 desarrollarán esquizofrenia.
No obstante, si Barres y Stevens tienen razón en que el sistema inmunológico es un mecanismo común detrás de los devastadores trastornos cerebrales, eso en sí mismo es un avance fundamental. Debido a que no conocemos los mecanismos que desencadenan tales enfermedades, los investigadores médicos solo han podido aliviar los síntomas en lugar de atacar las causas. No hay medicamentos disponibles para detener o incluso retardar la neurodegeneración en enfermedades como el Alzheimer. Algunas drogas elevan los neurotransmisores de manera que brevemente facilitan que las personas con demencia formen nuevas conexiones sinápticas, pero no reducen la velocidad a la que se destruyen las sinapsis existentes. Del mismo modo, no existen tratamientos que aborden las causas del autismo o la esquizofrenia. Incluso retrasar el progreso de estos trastornos sería un gran avance. Finalmente podríamos perseguir enfermedades que han estado sin control durante generaciones.
Estamos muy lejos de una cura, dice Stevens. Pero definitivamente tenemos un camino por delante.
Adam Piore es un escritor independiente que escribió Una forma impactante de arreglar el cerebro en noviembre/diciembre de 2015.