211service.com
La teoría matemática de la comunicación de Shannon aplicada a la secuenciación del ADN
Uno de los grandes héroes anónimos de la ciencia del siglo XX es Claude Shannon, un ingeniero de los famosos Laboratorios Bell durante su apogeo a mediados del siglo XX. La contribución más duradera de Shannon a la ciencia es la teoría de la información, que sustenta toda la comunicación digital.
En un famoso artículo que data de finales de la década de 1940, Shannon planteó el problema fundamental de la comunicación: reproducir, en un punto del espacio, un mensaje que se ha creado en otro. El mensaje primero se codifica de alguna manera, se transmite y luego se decodifica.
Shannon mostró que un mensaje siempre se puede reproducir en otro punto del espacio con precisión arbitraria, siempre que el ruido esté por debajo de algún nivel de umbral. Continuó calculando cuánta información podría enviarse de esta manera, una propiedad conocida como la capacidad de este canal de información.
Las ideas de Shannon se han aplicado ampliamente a todas las formas de transmisión de información con mucho éxito. Una vía particularmente interesante ha sido la aplicación de la teoría de la información a la biología: la idea de que la vida misma es la transmisión de información de una generación a la siguiente.
Ese tipo de pensamiento es continuo, revolucionario y aún se encuentra en sus primeras etapas. Hay mucho por venir.
Hoy, observamos un corolario interesante en el área de la transmisión de información biológica. Abolfazl Motahari y sus amigos de la Universidad de California, Berkeley, utilizan el enfoque de Shannon para examinar la rapidez con la que se puede extraer información del ADN mediante el proceso de secuenciación rápida.
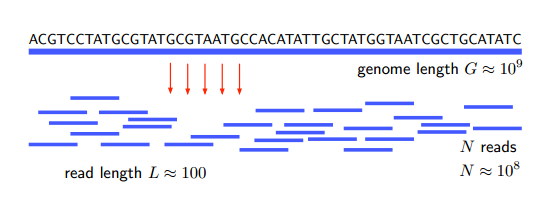
El problema aquí es determinar la secuencia de nucleótidos (A, G, C y T) en un genoma. Eso lleva mucho tiempo porque los genomas tienden a ser largos; por ejemplo, el genoma humano consta de unos 3 mil millones de nucleótidos o pares de bases. Esto llevaría una eternidad secuenciar en serie.
Entonces, el enfoque de la escopeta implica cortar el genoma en piezas aleatorias, que constan de entre 100 y 1,000 pares de bases, y secuenciarlas en paralelo. Luego, la información se vuelve a pegar en silico por un llamado algoritmo de reensamblaje.
Por supuesto, no hay forma de saber cómo reensamblar la información de una sola lectura del genoma. Entonces, en el enfoque de la escopeta, este proceso se repite muchas veces. Debido a que cada lectura divide el genoma de una manera diferente, las piezas inevitablemente se superponen con segmentos de una ejecución anterior. Estas áreas de superposición permiten volver a ensamblar todo el genoma, como un rompecabezas.
Eso huele a un problema clásico de la teoría de la información y, de hecho, varias personas han pensado de esta manera. Sin embargo, Motahari y compañía van un paso más allá al reafirmarlo más o menos exactamente como un análogo del famoso enfoque de Shannon.
Dicen que el problema de la secuenciación del genoma es esencialmente reproducir un mensaje escrito en ADN, en un formato electrónico digital. En este enfoque, el mensaje original está en ADN, se codifica para su transmisión mediante el proceso de lectura y luego se decodifica mediante un algoritmo de reensamblaje para producir una versión electrónica.
Lo que demuestran es que existe una capacidad de canal que define una tasa máxima de flujo de información durante el proceso de secuenciación. Proporciona el número máximo de pares de bases de ADN que se pueden resolver por lectura, mediante cualquier algoritmo de ensamblaje, sin tener en cuenta las limitaciones computacionales, dicen.
Ese es un resultado significativo para cualquier persona interesada en secuenciar genomas. Una pregunta importante es qué tan rápido una tecnología de secuenciación en particular puede hacer su trabajo y si es más rápida o más lenta que otros enfoques.
Eso no es posible de resolver en este momento porque muchos de los algoritmos utilizados para el ensamblaje están diseñados para tecnologías y enfoques de lectura específicos. Motohari y compañía dicen que hay al menos 20 algoritmos de reensamblaje diferentes, por ejemplo. Esto hace que sea difícil comparar diferentes algoritmos, dicen.
En consecuencia, nadie sabe realmente cuál es el más rápido o incluso cuál tiene el potencial de ser más rápido.
El nuevo trabajo cambia esto. Por primera vez, debería ser posible determinar qué tan cerca se acerca una tecnología de secuenciación dada al límite teórico.
Eso bien podría obligar a eliminar la madera muerta de esta área y estimular un período de rápida innovación en la tecnología de secuenciación.
Ref: arxiv.org/abs/1203.6233 : Teoría de la información de la secuenciación del ADN